







Nuclear receptors are ligand-inducible transcription factors that have been extensively studied in order to decipher the mechanisms of transcriptional regulation. They share a common organization in three main functional domains that coincide with homology regions. A central DNA binding domain corresponding to the highly conserved region C (10, 29) is flanked on either side by two transcriptional activation regions, AF1 and AF2.
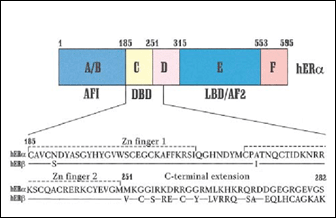 |
Fig. 1: Primary structure of estrogen receptors (from 29). AF1/2: activation function 1; DBD: DNA binding domain.
The DNA binding domain is composed of two zinc fingers (C4 type) that fold into a unique structural unit. Interactions with the phosphate backbone allow precise positioning of an a-helix in the major groove of the DNA response element, leading to formation of specific amino acid base pair interactions (2, 29).
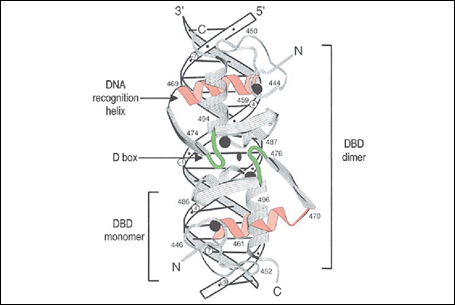 |
Fig. 2: Complex between the ER alpha DNA binding domain and a consensus ERE (from 29 and refs within).
Most nuclear receptors bind DNA as dimers, whether homodimers (steroid receptors) or heterodimers with an RXR partner (7, 12). Dimerization increases affinity and provides an additional element of specificity. The spacing between the repeated motifs in the response element is determined by the dimerization interface in the DBD, which involves different regions depending on the type of response element. Naturally-occurring mutations in the DBDs of nuclear receptors can lead to hormonal insensitivity, and can sometimes affect the selectivity of DNA recognition (28).
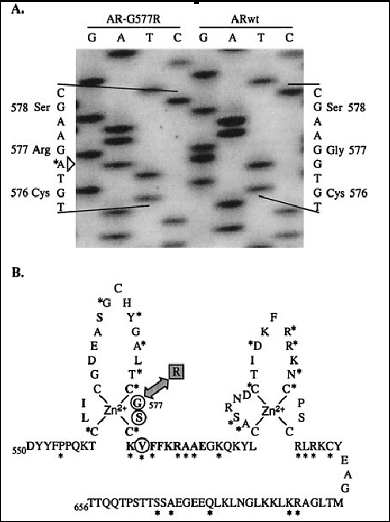 |
Fig. 3: The G577R mutation in the androgen receptor affects a residue important for DNA binding site discrirmination (from 28)
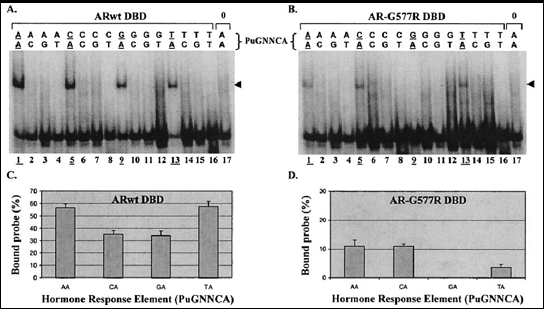 |
Fig. 4: The wild-type and mutant AR DBDs recognize DNA elements with different affinities and specificities (from 28)
Nuclear response elements have been characterized traditionally through promoter analysis (1, 3, 6, 13). However, whole-genome screens for high affinity estrogen response elements have suggested that these elements likely outnumber receptor molecules in target cells (39). Although only a small proportion of these elements appears conserved between the human and mouse genomes, interaction of estrogen receptors with non-conserved elements can be observed in chromatin immunoprecipitation experiments (39). Questions that remain to be addressed are whether access to binding sites is modulated in a tissue-specific manner, and how the position of the response element in gene flanking sequences (proximal vs distal) affects cofactor recruitment.
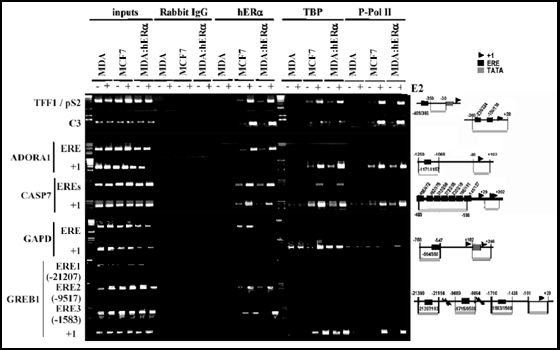 |
Fig. 5: Chromatin immunoprecipitation assays performed with antibodies against the estrogen receptor on EREs identified through bioinformatics screening of the human genome (from 39).
DNA-bound nuclear receptors recruit transcriptional coactivators, usually in a ligand-dependent manner. Coactivators can recruit the basal transcription machinery to form pre-initiation complexes on the DNA, or facilitate access of transcription factors to promoter sequences by chromatin modifying/remodeling activities. The ligand-binding domain of nuclear receptors contains a transcriptional activation domain (activating function AF2), which recruits a number of coactivators through a common binding interface (22). Corepressors can also be recruited in a ligand-dependent manner (34).
Fig. 1: The typical organization of nuclear receptors in 6 homology regions is shown, as well as the main functions associated with each region. The two transcriptional activation domains (AF1 and AF2) are indicated by bars, and recruitment of coactivators through these domains is schematized. Note that several other coactivators and corepressors have been shown to interact with ERalpha.
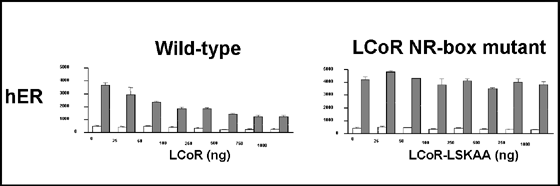 |
Fig. 2: LCoR is an estrogen receptor corepressor containing LXXLL motifs (from 34).
Antiestrogens are estrogen receptor antagonists that have been developed to inhibit the growth stimulatory activity of estrogen on breast cancer cells (Fig. 3). Antiestrogens bind ERs in a manner similar to estrogen, but induce a different conformation of the ligand binding domain due to the presence of a lateral chain grafted onto a steroid or steroid-like skeleton (Fig. 3). This results in lack of recruitment of coactivators by the AF2 region. However, some antiestrogens act as partial agonists in a tissue-specific manner. In particular, tamoxifen and raloxifene can display agonist activity in uterine cancer cells both at the level of transcriptional regulation and tumor growth (19, 31).
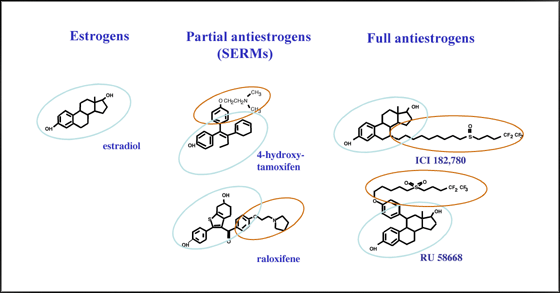 |
Fig. 3: Structure of estrogen receptor agonists and antagonists.
A number of mutations can modluate the agonist activity of antiestrogens. Tamoxifen and raloxifene both contain in their side chains a tertiary amine that makes contact with a charged amino acid in the LBD, D351. Mutagenesis of position 351 indicated that this interaction is not crucial for the affinity of receptor-antiestrogen interaction and is not required for the antagonist activity of tamoxifen (24). However, mutations that abrogate the charge of D351 abolish the agonist activity of tamoxifen in permissive cell systems, suggesting a role of amino acid D351 in modulating protein-protein interactions that mediate agonist activity. Other mutations under study in the laboratory confer increased agonist activity to raloxifene and/or ICI182,780. Our studies are aimed at characterizing both the structural and functional alterations resulting from these mutations using protein modeling as well as conformational and functional assays of estrogen receptors.
Retinoic acid is synthesized in target cells via a two-step oxydation of retinol precursors, involving retinol and retinal dehydrogenases. We have been investigating the role of retinal dehydrogenase RALDH1 in retinoic acid synthesis and the molecular basis for its specificity for retinal substrates, in collaboration with the laboratory of Dr. P.V. Bhat (Hopital Hotel-Dieu de Montréal). Rat RALDH1 is abundant in adult kidney, and its expression patterns during post-natal kidney development are suggestive of a potential role in tubulogenesis (21).
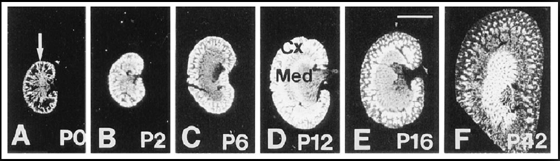 |
Fig. 1: Expression patterns of rat RALDH during post-natal kidney development (from 21).
The rat RALDH1 promoter is sufficient to drive expression in kidney cells (33). We have also cloned the monkey RALDH1 gene and observed a role of the endogenous enzyme in the synthesis of retinoic acid in monkey proximal tubule cells; this enzyme is active with all-trans, 9-cis and 13-cis retinal, whereas rat RALDH1 is only active with the all-trans and 9-cis isoforms (35). Finally, we have identified amino acids in RALDHs involved in substrate specificity for retinal isomers and obtained chimeric enzymes specific for either all-trans or 9-cis retinoic acid (32).
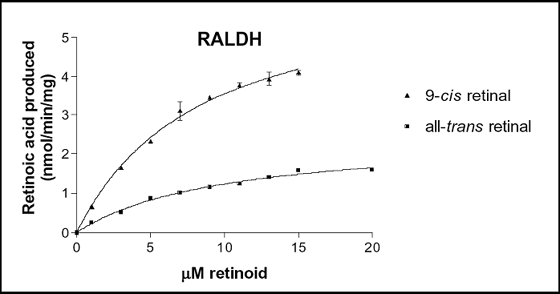 |
Fig. 2: Enzymatic properties of wild type RALDH
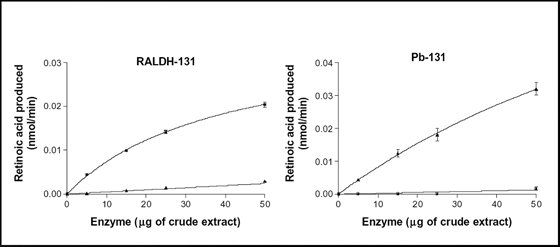 |
Fig. 3: Enzymatic properties of chimeras RALDH-131 and Pb-131